| |
A female infant with hypotonia, developmental delay, transitional hearing loss and 22q13.1 deletion
Chulaluck Techakittiroj, Hans Andersson, Kelly Jackson, Chris Dvorak and Marilyn Li
New Orleans, USA
Author Affiliations: Hayward Genetics Center, Tulane University School of Medicine (Techakittiroj C, Andersson H, Jackson K, Dvorak C and Li M); Department of Pediatrics, Tulane University School of Medicine (Techakittiroj C, Andersson H, Jackson K and Li M), New Orleans, LA, USA
Corresponding Author: Marilyn M. Li, MD, Hayward Genetics Center 1430 Tulane Ave. SL-31, New Orleans, LA 70112, USA (Tel: 1-504-988-5101; Fax: 1-504-988-1763; Email: mli2@tulane.edu)
This article was presented at the International Congress of Global Chinese Geneticists 2006 (ICGCG 2006).
Chromosome 22q13 deletion syndrome (MIM 606232) is associated with global developmental delay, generalized hypotonia, absent or severely delayed speech, normal to accelerated growth, and specific facial features. We report a female infant with persistent hypotonia, transitional hearing loss, developmental delay, specific facial features, and a terminal deletion of chromosome 22q13.1. Parental chromosomal studies demonstrated that the deletion was de novo. Most clinical features of the patient were similar to those of the previously reported patients with 22q13 deletion, but some of them such as hypoplastic toenails were unique. The phenotype variation in patients with 22q13 deletion could be attributed to different deletions that vary in size and involve overlapping but diverse genomic segments.
Key words: 22q13 deletion; developmental delay; hypotonia; hearing loss; FISH
World J Pediatr 2006;4:303-307
Introduction
Chromosome 22 involves many well defined genetic disorders. There are at least two deletion syndromes linked to chromosome 22q: 22q11.2 deletion associated with DiGeorge syndrome and Velocardiofacial syndrome, and 22q13 deletion syndrome characterized by generalized developmental delay, hypotonia, severely compromised language development, and specific facial features. 22q13 deletion syndrome was first described by Watt and his colleagues in 1985.[1] Since then there have been many cases of 22q13 deletion reported. In 2001, Phelan et al[2] studied and reviewed clinical features of 61 patients with 22q13.3 deletion and summarized the common clinical features of the syndrome. The common clinical features include severe expressive language delay, mild mental retardation and minor dysmorphism (prominent or dysplastic ears, syndactyly, epicanthal fold, etc). Since then, more cases of 22q13 deletion have been reported.[3,4] The reported patients shared similar clinical features described in Phelan's review, although phenotype variation was noted. In this report, we reported a patient with 22q13.1 deletion and compared her clinical features with those of the reported 22q13 deletion syndrome.
Case report
The patient was referred for genetic evaluation at two months of age. She was noticed to have severe hypotonia, mild dysmorphic facial features including bilateral epicanthal folds, depressed nasal bridge, and almond shaped eyes (Fig. 1), redundant neck skin, bilateral hearing loss, heart murmur, hypoplastic toenails (Fig. 2), and swollen labia majora. Echocardiography showed an open ductus arteriosus. Otoacoustic emission (OAE) test revealed bilateral absence of emissions at birth. Auditory brainstem response (ABR) displayed moderate hearing loss at 500 Hz and from 2000 to 4000 Hz at 6 weeks old. The patient was born to a 22-year-old G2P0 mother at 41 weeks of gestational age by cesarean section. At birth, her weight was 7 lb. 11 oz (50-75 percentile), her height was 19.5 inches (50-75 percentile), and her head circumference was 14.75 inches (>90 percentile). The pregnant history was unremarkable except breach position, which resulted in cesarean section delivery. The mother had 1 miscarriage at 2 and a half months gestational age due to blighted ovum. Family history revealed that the father was born with undescended testes, and a maternal uncle had learning disability. There were several distant relatives with "mental retardation" and one relative with "Turner syndrome". Ethnic background was French, German and Italian. There was no history of consanguinity. At 4 months follow-up, she continued to have severe hypotonia with no head control. Her audio capability was slightly improved. At 10-month follow-up, she was still hypotonic and could not sit up without support. However, the results of her hearing tests, both OAE and ABR, were within normal range. Growth parameters at 10 months old were at the same level as those at birth: height at 90%, weight and head circumference at 75%.
Peripheral blood from the patient and her parents was cultured with phytohemagglutinin (PHA) stimulation for 72 hours before harvest. Chromosome preparation and G-banding were carried out using the standard protocols of Tulane Cytogenetics Laboratory. At least 20 high resolution metaphase spreads were analyzed for each sample. Images were captured and karyotyped using CytoVision™ Capture & Karyotyping Workstation (Applied Imaging Corp., San Jose, CA, USA).
Chromoprobe Multiprobe® - T System (Cytocell Technologies, Cambridge, England) consisting of 44 subtelomere probes for the short and long arm of chromosome X, Y, 1 to 12, 16 to 20 and long arms of chromosome 13 to 15 and 21 to 22 was used to determine whether the deletion was terminal or interstitial, and to rule out unbalanced cryptic translocation. DNA probes ARSA and TUPLE1 specific for chromosome locations 22q11.2 and 22q13, respectively (Vysis, Downer Grove, IL) were used to further define the deletion breakpoint. Fluorescence in situ hybridization (FISH) studies were performed according to the manufacturer's protocols with minor modification. Five metaphases and 20 interphase nuclei were analyzed for each subtelomere probe, 10 metaphases were analyzed for probes ARSA and TUPLE1. FISH images were captured and analyzed using filter-based imaging system CytoVision ChromoFluor™ (Applied Imaging Corp., San Jose, CA).
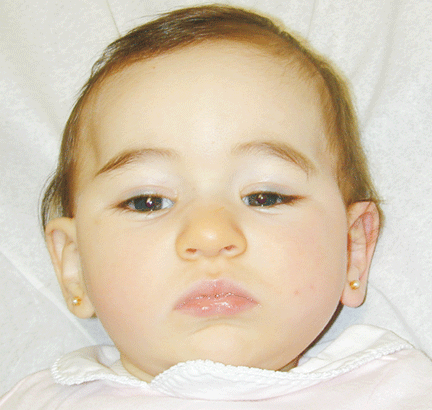 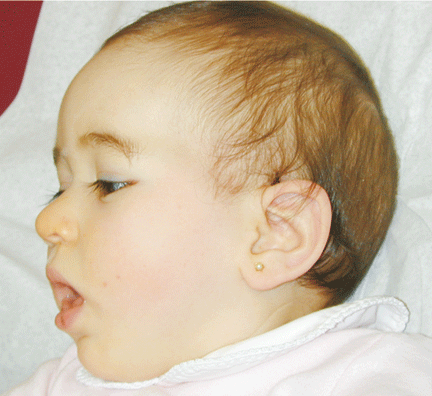 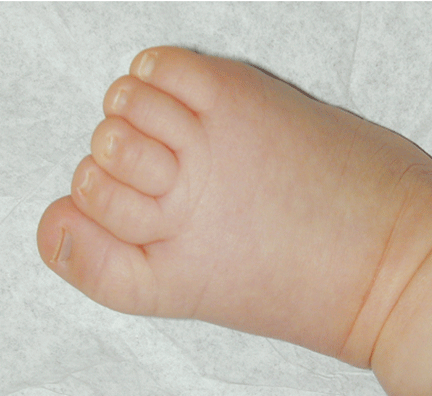
Fig. 1. Front and side views of the patient at 10-month age revealed bilateral epicanthal folds, depressed nasal bridge, and almond shaped eyes.Fig. 2. Right foot showing hypoplastic and convex toenails.

Fig. 3. Cytogenetic study showing a deleted chromosome 22 (arrow) in the patient.
 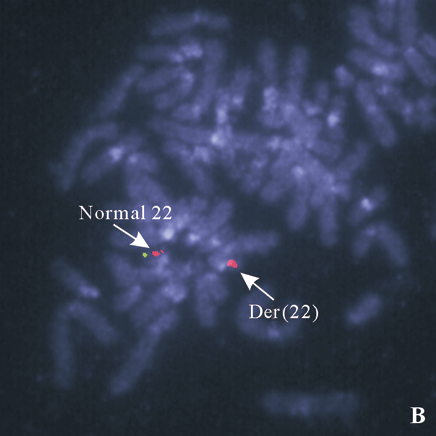
Fig. 4. A: 22q subtelomere probe showing one signal on the normal 22, no signal on the del(22); B: a deletion of ARSA (green) probe on the der(22) shown by FISH with probe ARSA and TUPLE1.
Table. Features of the patient with 22q13 deletion
|
|
Present case |
Prasad et al[4] |
Fujita et al[5] |
Romain et al[4] |
Luciani et al[7] |
Phelan et al[2] |
|
Karyotype |
46XX, ish del
(22)(q13.1) |
46XY, del (22)(q13.1) |
46,XX ish del (22)(q13.1q13.2) |
q13.1-q13.33 |
|
|
|
Developmental delay |
+ |
+ |
+ |
+ |
100% |
92%-100% |
|
Speech delay |
N/A |
|
+ |
+ |
100% |
91%-100% |
|
Hypotonia |
+ |
+ |
+ |
+ |
82% |
74%-97% |
|
Birth weight |
50-75th percentile |
>90th percentile |
AGA |
AGA |
|
|
|
Postnatal growth |
75th percentile |
95th percentile |
75-90th percentile |
N |
N to accelerated growth |
N to accelerated growth |
|
Hearing loss |
partial |
- |
+ |
± |
|
|
|
Dolicocephaly |
- |
N/A |
+ |
- |
|
39%-57% |
|
Eyes
ptosis
epicanthal fold |
-
+ |
deep set
|
+
+ |
+
+ |
|
13%-57%
38%-41% |
|
Prominent/
dysplastic ears |
N |
prominent |
dysplastic |
dysplastic |
82% |
47%-65% |
|
Philtrum |
N |
smooth |
long |
long |
|
|
|
High arched palate |
- |
+ |
+ |
- |
|
|
|
Nails |
hypoplastic |
hypoplastic |
N/A |
N/A |
|
|
|
Syndactyly |
- |
- |
- |
- |
|
28%-38% |
|
CVS |
small PDA |
N |
N |
N/A |
|
|
|
Chest wall |
N |
mild pectus excavatum |
N/A |
N/A |
|
|
N: normal; N/A: not applicable; CVS: chorionic villi sampling; PDA: patent ductus arteriosus.
Results
High resolution chromosomal studies showed a karyotype of 46XX, del(22)(q13.1) or der(22)t(?;22) for the patient (Fig. 3). Chromosomal studies on the parents showed normal results.
FISH for subtelomere probes demonstrated only one copy of the 22q subtelomere probe was present. No missing or gaining signals were observed for other 43 telomere probes in the specimen. These results indicated that the patient carried a terminal deletion on one copy of chromosome 22. FISH studies with ARSA and TUPLE1 probes showed two signals for the TUPLE1 probe and only one signal for the ARSA probe. These results indicated that the breakpoint of the deleted 22 was proximal to the ARSA locus at 22q13.3.
Discussion
We reported a female patient with hypotonia and dysmorphic features. Cytogenetic study showed a deletion of the long arm of one chromosome 22. The cytogenetic deletion could be terminal or interstitial. It could also be the result of an inter-chromosomal rearrangement (cryptic translocation), in which the terminal segment including subtelomeric region of the aberrant 22q is replaced by a terminal segment from another chromosome. To further delineate the abnormal chromosome 22, we performed FISH with 44 subtelomere probes. Chromosome 22q subtelomere probe hybridized to one homolog of chromosome 22 but not the other (Fig. 4A). All other subtelomere probes showed normal hybridization pattern. These results demonstrated that the abnormal chromosome 22 was a deleted chromosome 22, and the deletion was terminal. High resolution chromosome analysis showed one dark band at the end of the long arm of the deleted chromosome 22, indicating the breakpoint of q13.1 (Fig. 3). To confirm the breakpoint, we performed FISH study with ARSA probe at 22q13.3 along with TUPLE1 as a control probe. The results showed two signals for the TUPLE1 probe and one signal for the ARSA probe, suggesting the breakpoint of 22q13.1 (Fig. 4B). Parental chromosome studies showed normal results; therefore, the del(22q13.1) in our patient was de novo. Compared with our patient (Table), only one patient in the literature had exactly the same terminal deletion as our patient.[4] The other 2 reported to have the same breakpoint were interstitial deletions.[5,6] In 2003, Luciani et al[7] and Wilson et al[8] published two separate but similar articles describing clinical features and molecular analysis results on 88 patients with 22q13 deletion. Common clinical features of the syndrome from their study included hypotonia, global developmental delay, expressive speech delay, prominent or dysplastic ears and behavior disorders, which are similar to those previously reported. Developmental delay was seen in all 22q13 patients, and hypotonia in those patients with a deletion exceeding 4 Mb.[7] The severity of phenotype correlates to the size of the deletion. In our patient, there were a big 22q deletion, severe hypotonia, and developmental delay, which were identical with published observations. A poor prognosis was considered for our patient since the size of her deletion was quite large (estimate 10 to 12 Mb). Compromised language development was observed essentially in all reported patients, but it was too early to have a meaningful assessment of our patient's language capability, although no sign of single word speech was noticed by her parents at 10 months of age. Hearing impairment was reported in one third of the patients. Our patient showed a transitional hearing loss, which could be missed in patients diagnosed at an older age. Another striking feature of our patient was her growth rate. She had been on 50-75 percentile for all growth parameters (height, weight, and head circumference) while both of her parents were at 10-25 percentile of adult height and weight. Over 80% of the reported patients with 22q13 deletion syndrome showed normal or accelerated growth. This is not commonly seen in patients with chromosomal abnormalities. A gene or genes that negatively control cell growth may reside in the commonly deleted region. It was reported that growth retardation was seen in all patients with ring chromosome 22 [r(22)], in which the chromosome segment commonly deleted in 22q13 deletion syndrome was also deleted. We propose that cell death caused by genetic instability, i.e., continuous structure change of r(22)chromosome and formation of micronuclei, may be responsible for growth retardation in r(22) syndrome.
Luciani et al[7] proposed that haploinsufficiency of PROSAP2/SHANK3, ACR, and RABL2B may be responsible for the phenotype of 22q13 deletion syndrome based on the observation of two patients with a 160 kb deletion immediately proximal to 22q telomere, and that genes deleted or interrupted in these patients include PROSAP2/SHANK3, ACR, and RABL2B. Studies from Wilson et al[8] suggested that haploinsufficiency of the gene SHANK3 is a major causative factor for the neurological symptoms of 22q13 deletion syndrome. SHANK3 is located within 200 kb of chromosome 22q telomere. In rats, SHANK proteins, including SHANK3, have been localized to the postsynaptic density of excitatory synapses, and are important in linking the metabotropic glutamate receptors through the interaction with a homer homodimer and NMDA receptors.[9] Because SHANK3 is involved in a continuous protein network in the postsynaptic density, alteration of the relative proportions of such a protein could alter synapse formation and function, making SHANK3 an excellent candidate for affecting neurological functions. However, Fujita et al[5] reported a patient with interstial deletion of 22q13.1 to 22q13.2 showing the same clinical features as other 22q13 deletion patients, indicating the involvement of other genes that may also influence neurological functions.
Acknowledgements
We are grateful to the parents of the patient and the referring physician for their support and cooperation.
Funding: None.
Ethical approval: Not needed.
Competing interest: None.
Contributors: No other contributors other than the authors.
References
1 Watt JL, Olson IA, Johnston AW, Ross HS, Couzin DA, Stephen GS. A familial percentric inversion of chromosome 22 with a recombinant subject illustrating a 'pure' partial monotony syndrome. J Med Genet 1985;22:283-287.
2 Phelan MC, Rogers RC, Saul RA, Stapleton GA, Sweet K, McDermid H, et al. 22q13 deletion syndrome. Am J Med Genet 2001;101:91-99.
3 Precht KS, Lese CM, Spiro RP, Huttenlocher PR, Johnston KM, Baker JC, et al. Two 22q telomere deletions serendipitously detected by FISH. J Med Genet 1998;35:939-942.
4 Prasad C, Prasad AN, Chodirker BN, Lee C, Dawson AK, Jocelyn LJ, et al. Genetic evaluation of pervasive developmental disorders: the terminal 22q13 deletion syndrome may represent a recognizable phenotype. Clin Genet 2000;57:103-109.
5 Fujita Y, Mochizuki D, Mori Y, Nakamoto N, Kobayashi M, Omi K, et al. Girl with accelerated growth, hearing loss, inner ear anomalies, delays myelination of the brain and del (22)(q13.1q13.2). Am J Med Genet 2000;92:195-199.
6 Romain DR, Goldsmith J, Cairney H, Columbano-green LM, Smythe RH, Parfitt RG. Partial monosomy for chromosome 22 in a patients with del(22)(pter→q13.1::q13.3→qter). J Med Genet 1990;27:588-589.
7 Luciani JJ, de Mas P, Depetris D, Mignon-Ravix C, Bottani A, Prieur M, et al. Telomeric 22q13 deletions resulting from rings, simple deletions, and translocations: cytogenetics, molecular, and clinical analyses of 32 new observations. J Med Genet 2003;40:690-696.
8 Wilson HL, Wong AC, Shaw SR, Tse WY, Stapleton GA, Phelan MC, et al. Molecular characterization of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J Med Genet 2003;40:575-584.
9 Boeckers TM, Bockmann J, Kreutz MR, Gundelfinger ED. ProSAP/Shank proteins―a family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J Neurochem 2002;81:903-910.
Received September 12, 2006 Accepted after revision September 16, 2006
|
