| |
Disorders of sex development: update on the
genetic background, terminology and risk for
the development of germ cell tumors
Martine Cools, Leendert HJ Looijenga, Katja P Wolffenbuttel, Sten LS Drop
Ghent, Belgium and Rotterdam, The Netherlands
Author Affiliations: Department of Pediatrics, Division of Pediatric Endocrinology, University Hospital Gent, De Pintelaan 185, 9000 Gent, Belgium (Cools M); Department of Pathology, Erasmus Medical Center, Josephine Nefkens Institute, Daniel Den Hoed Cancer Clinic, Postbus 2040, 3000 DR Rotterdam, The Netherlands (Looijenga LHJ); Department of Urology (Wolffenbuttel KP) and Department of Pediatrics (Drop SLS), Sofia Children's Hospital, Dr Molewaterplein 60, 3015 GJ Rotterdam, The Netherlands
Corresponding Author: Martine Cools, Department of Pediatrics, Division of Pediatric Endocrinology, University Hospital Gent, Building 5K6, De Pintelaan 185, 9000 Gent, Belgium (Tel: +32 93324728; Email: martine.cools@ugent.be)
doi:10.1007/s12519-009-0020-7
Background: Considerable progress has been made on genetic mechanisms involved in disorders of sex development and on tumor formation in dysgenetic gonads. Clinical and psychological outcome of patients are, as far as evaluated, unsatisfactory at present. Guidelines are emerging in order to optimize long-term outcome in the future.
Data sources: The information obtained in this review is based on recent original publications and on the experience of our multidisciplinary clinical and research group.
Results: This review offers an update on our knowledge concerning gene mutations involving in disorders of sex development, on the renewed nomenclature and classification system, and on the mechanisms of tumor development in patients.
Conclusions: The consensus meeting on disorders of sex development has renewed our interest in clinical studies and long-term outcome of patients. Psychological research emphasizes the importance to consider male gender identity wherever possible in cases of severe undervirilization. Patient advocacy groups demand a more conservative approach regarding gonadectomy. Medical doctors, scientists and governmental instances are increasingly interested in the set-up of international research collaborations. As a consequence, it is expected that new guidelines for the optimal care of patients will be proposed in the coming years.
Key words: consensus; germ cell tumor; nomenclature; risk; sex development
World J Pediatr 2009;5(2):93-102
Introduction
Disorders of sex development (DSD) is regarded as an umbrella term, referring to a group of congenital conditions in which the development of the chromosomal, gonadal or anatomical sex has been atypical.[1,2] DSD has replaced the formerly used term "intersex".
DSD represents a markedly heterogeneous group of conditions. On one hand, identical genetic defects may result in various clinical presentations; on the other hand, a similar phenotype may be the end point of different genetic pathways. A detailed insight in the stepwise normal process of sex development and the different genes involved is essential to understand the mechanisms eventually leading to DSD.
Specific subgroups of DSD patients are prone to the development of a germ cell tumor (GCT). Their individual risk profile can be estimated by combining knowledge on the specific gonadal differentiation patterns present in patients, and detailed histological and genetic diagnostic investigations.
The process of normal sex development
The embryonic precursors of the gonads and genital structures are bipotential, in other words, they have the potential to differentiate either into the male or female direction, depending on the correct and timely expression of specific genes. In mammals, male sex differentiation is initiated by the expression of SRY, leading to a cascade of genetic expression mechanisms, and finally resulting in the full masculinization of internal and external genital structures. In case SRY is not (or not correctly) activated, the primitive genital structures have an intrinsic tendency to differentiate into the female direction.[3] In summary, the genetic sex (the presence or absence of the Y chromosome) determines the gonadal sex (differentiation of the bipotential gonad into the testis or ovary). The fetal testis produces hormones (testosterone (T) and anti-Müllerian hormone (AMH)), resulting in masculinization of the internal and external genital strucutres (male phenotypic sex). In the absence of (sufficient action of) these hormones, the phenotypic sex of the individual will be female.[3,4]
Gonadal sex: formation of the bipotential gonad, gonadal differentiation and the origin of gonadal dysgenesis
The bipotential gonad develops from the urogenital ridge in close association with the future kidney and adrenal. In humans, several genes related to this process have been identified, namely WT1, SF1 and DAX1. A mutation in one of these genes does not only result in gonadal dysgenesis (GD), but is often associated with specific pathology of the kidney (Danys-Drash syndrome, Frasier syndrome, WAGR syndrome in case of WT1 mutation) or the adrenal (congenital adrenal hypoplasia in case of SF1 or DAX1 mutation) (Tables 1, 2).
Initially, the undifferentiated gonad does not contain any germ cells; they are set apart in the the yolk sac, from which they migrate and disseminate over both gonads during the fourth and fifth weeks of pregnancy.[9]
During the sixth week, SRY, and immediately thereafter also SOX9 are expressed in the presence of a Y chromosome, under the influence of specific regulatory genes (SF1 and WT1 among others).[3] The activated SRY-SOX9 tandem is at the top of a cascade of genetic reactions, controlling the gradual differentiation of the gonad, first into a primitive testis with sex cords (primitive organization of germ cells and polarized Sertoli cells, characterized by SOX9 activation), then into a more mature testis with seminiferous tubules.[10] A rigorous time schedule, a continuous balance between activating and inhibiting mechanisms, and a correct dosage of gene products are essential in this process: due to the cascade effect, a minor aberration can have major consequences for testis development.[3-5]
For a long time it was believed that the absence of SRY automatically leads to full ovarian differentiation, characterized by the initiation of meiosis of the germ cells and the organization of germ cells and granulosa cells into primordial follicles. However, at present, a number of genes essential to this process have been identified, such as WNT4, FOXL2 and RSPO1[3,4,11] so the process of ovarian differentiation is no longer considered as the default pathway. The finding of streak gonads and primitive sex cords instead of ovarian follicles in 46, XY (SRY negative) females is in line with this hypothesis (personal observations).
Recently, a mouse model was proposed in which the decision towards testicular or ovarian differentiation depends on the balance between two opposing signals: in the bipotential gonad, FGF9 and WNT4 are equally expressed. SOX9 induces upregulation of FGF9, which leads to an imbalance between FGF9 and WNT4, and thus testis formation is initiated. In the absence of SOX9 (in the XX gonad), WNT4 is upregulated and ovarian differentiation is initiated.[12] Whether this model is correct and applicable to the human gonad remains to be elucidated.
Phenotypic sex: hormonal disturbances at the origin of under- and overvirilization syndromes
In the absence of testicular hormones, Müllerian structures differentiate into tubae uterinae, uterus and the upper part of the vagina. From the urogenital sinus, the lower vagina, the clitoris and the labiae minorae are formed.
Hormonal production by the primitive testis is initiated in early pregnancy: from the ninth week of pregnancy onwards, Sertoli cells produce AMH, leading to regression of the Müllerian ducts.[10,13] Inhibin B, another Sertoli cell product, is important for germ cell-Sertoli cell interaction.[14] Shortly thereafter, Leydig cells start their hormonal production: insulin-like factor 3 (INSL3) is involved in the transabdominal phase of testicular descent,[15] while T leads to masculinization of internal and external genital structures: the Wolffian ducts develop into epididymis, vas deferens and seminal vesicles, and the urogenital sinus differentiates into the prostate, scrotum and penis with the meatus urethrae on top of it. In contrast to the internal genital structures, the urogenital sinus is not sensitive to T itself, but to its metabolite dihydrotestosterone (DHT), which is formed locally under the influence of the enzyme 5α-reductase.[4]
At present, one gene is known to be involved in Müllerian duct development in humans: a mutation in the WNT4 gene has been associated with the Mayer-Rokitanski syndrome (congenital absence of tubae, uterus and upper part of the vagina), in association with hyperandrogenism.[16] In mice, the involvement of several other genes has been described.[13]
Androgen synthesis takes several steps (Fig. 1). A defect in the production (Smith-Lemli-Opitz syndrome) or transport (steroidogenic acute regulatory protein (StAR) deficiency) of its precursor cholesterol results in severe undervirilization (besides other characteristics), as well as a mutation in the gene encoding the LH/HCG receptor (Leydig cell hypoplasia).
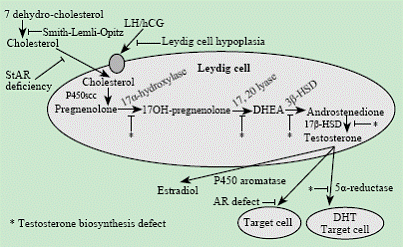 Fig. 1. Schematic overview of androgen biosynthesis in the testis. Androgen biosynthesis is a stepwise process, starting from cholesterol. In the Smith-Lemli-Opitz syndrome, the last step in cholesterol synthesis (the formation of cholesterol from 7-dehydrocholesterol) is disturbed. A defect in the transport of cholesterol is caused by the absence of its transportation protein (steroidogenic acute regulatory protein (StAR) deficiency). Testosteron biosynthesis is initiated after LH stimulation, which binds to the Leydig cell via its LH/HCG receptor. Several enzymes are involved in androgen biosynthesis (cholesterol side chain cleavage, 17α-hydroxylase, 3β hydroxysteroid dehydrogenase type II, 17β hydroxysteroid dehydrogenase type III) as well as the P450 oxidoreductase, a necessary cofactor for the P450 enzyme complex. Testosteron is converted into dihydrotestosteron at the target cell via the enzyme 5α-reductase. Both hormones act through binding to the androgen receptor. Fig. 1. Schematic overview of androgen biosynthesis in the testis. Androgen biosynthesis is a stepwise process, starting from cholesterol. In the Smith-Lemli-Opitz syndrome, the last step in cholesterol synthesis (the formation of cholesterol from 7-dehydrocholesterol) is disturbed. A defect in the transport of cholesterol is caused by the absence of its transportation protein (steroidogenic acute regulatory protein (StAR) deficiency). Testosteron biosynthesis is initiated after LH stimulation, which binds to the Leydig cell via its LH/HCG receptor. Several enzymes are involved in androgen biosynthesis (cholesterol side chain cleavage, 17α-hydroxylase, 3β hydroxysteroid dehydrogenase type II, 17β hydroxysteroid dehydrogenase type III) as well as the P450 oxidoreductase, a necessary cofactor for the P450 enzyme complex. Testosteron is converted into dihydrotestosteron at the target cell via the enzyme 5α-reductase. Both hormones act through binding to the androgen receptor.
Mutations are described in every single gene involved in androgen biosynthesis (P450scc, P450c17, 3βHSDII, 17βHSDIII, 5α-reductase), and in the gene encoding the enzyme P450 oxidoreductase (POR), functioning as an indispensable cofactor for the P450 enzyme complex.[17,18] At birth, the resulting phenotype for all of these conditions is highly variable from normal female to pronounced genital ambiguity. Two conditions, namely 17β-hydroxysteroid dehydrogenase (17β-HSD) deficiency and 5α-reductase deficiency, are characterized by pronounced virilization during puberty, presumably due to the activation of iso-enzymes that are inactive during fetal life. In all cases of 46, XY undervirilization, the uterus is absent due to the preserved action of AMH (the defect only concerns T synthesis or its bioactivity).
The androgen insensitivity syndrome (AIS) is characterized by partial (partial AIS, PAIS) or no androgen action (complete AIC, CAIS) at all, in spite of a normal or elevated T production. This is due to a mutation in the androgen receptor and results in an unambiguously female phenotype with abdominal, inguinal or labial testicles in CAIS, whereas in PAIS, a variety of clinical phenotypes is possible, depending on the degree of residual androgen activity.[19,20] The gene encoding the androgen receptor is located on Xq containing 8 exons. In all of them, several mutations have been demonstrated, and genotype-phenotype variation has been described within the same families affected by PAIS.[21] Female carriers have no symptoms but can pass the gene defect to their offspring.
During fetal life, virilization can occur due to excessive androgen production by the female fetus itself, by the mother, or by the placenta, causing the development of a (micro) penis, labial fusion and insufficient outgrowth of the vesicovaginal septum, which leads to the presence of a urogenital sinus, mostly in a hypospadic position. Excessive fetal androgen production is mostly due to mutations in genes encoding enzymes involved in steroidogenesis (P450c21, P450c11β), and results in congenital adrenal hyperplasia (CAH).[17] Increased androgens are produced by the placenta in the rare case of fetoplacental aromatase deficiency, which is due to a defect in the P450aro gene, responsible for the conversion of T into estradiol at the fetal and the placental level. This defect causes increased androgen levels and virilization in both the fetus and its mother.[17] In 46, XX DSD overvirilized patients, the internal genital structures are always female (including the presence of a uterus): functional ovaries are present, and there is no AMH production, allowing normal development of the Müllerian structures, whereas the Wolffian ducts disappear since they are only sensitive to the paracrine action of T (thus, Wolffian structures do not develop under the influence of increased T levels from adrenal origin). The different genes involved in the development of undervirilization or overvirilization are summarized in Table 3.
Classification of DSD
Several classification systems have been developed in the past to offer a comprehensive overview of DSD.[4,24] However, none of them has managed to summarize the different forms of DSD in an unambiguous way. This was in part due to the existing nomenclature designating the various disorders: terms such as "intersex", "male or female pseudohermaphroditism", "hermaphrodite", "dysgenetic male pseudohermaphroditism" are not specific, confusing and are experienced as stigmatizing and pejorative by several patients. In October 2005, an international consensus meeting was organized with regard to DSD, bringing together mainly specialists from the European, American and Australian continents. One of the major topics at this conference was a revision of the existing nomenclature, which had to become more neutral and less confusing and which preferably had to refer to the genetic background of DSD. This resulted in the proposal of a new classification system, as presented in Table 2.[1,2]
Table 1. Overview of genes involved in the formation of the bipotential gonad and the process of gonadal differentiation[1-8]
|
Condition
|
Gene
|
Uterus
|
Adrenal pathology
|
Frequently associated characteristics
|
|
46, XY DSD with GD
|
|
WAGR, Danys-Drash, Frasier
|
WT1
|
+/-
|
-
|
Proteïnuria, Wilms' tumor, renal abnormalities
|
|
Steroidogenic factor 1 (SF1)
|
NR5A1
|
+/-
|
+/-
|
+/- Partial hypogonadotropic hypogonadism
Mutation can also lead to an isolated androgen biosynthesis
defect or PAIS-like condition
|
|
Xp21 duplication
|
DAX1
|
+/-
|
-
|
Duplication: incomplete testis differentiation
Mutation/deletion: congenital adrenal hypoplasia
|
|
SRY
|
SRY
|
+/-
|
-
|
-
|
|
SOX9
|
SOX9
|
+/-
|
-
|
Campomelic dysplasia
|
|
Desert hedgehog
|
DHH
|
+
|
-
|
+/- Minifascicular neuropathy
|
|
X-linked lissencephaly
|
ARX
|
-
|
-
|
Lissencephaly, epilepsy
|
|
9p24.3 deletion
|
DMRT1
|
+/-
|
-
|
Mental retardation
|
|
SIDDT syndrome
|
TSPYL1
|
-
|
-
|
Sudden infant death
|
|
Xq13.3 deletion
|
ATRX
|
-
|
-
|
α-thalassemia, mental retardation
|
|
1q35 duplication
|
WNT4
|
+/-
|
-
|
WNT4 deletion in 46,XX: hyperandrogenism, Mayer-
Rokitanski |
|
46, XX DSD with GD
|
|
SRY translocation
|
SRY
|
+/-
|
-
|
-
|
|
SOX9 duplication
|
SOX9
|
+/-
|
-
|
-
|
|
Palmo-plantar hyperkeratosis
|
RSPO1
|
+/-
|
-
|
Palmar and plantar hyperkeratosis, squammous cell carcinoma
|
DSD: disorder of sex development; GD: gonadal dysgenesis; WAGR: Wilms' tumor aniridia genitourinary anomalies mental retardation; PAIS: partial androgen insensitivity syndrome.
Table 2. New classification of DSD[1,2]
|
Sex chromosomal DSD
|
46, XY DSD
|
46, XX DSD
|
|
45, X (Turner syndrome and variants)
47, XXY (Klinefelter syndrome and variants)
45, X/46, XY and variants
46, XX/46, XY and variants |
Gonadal (testicular) dysgenesis
1. complete GD
2. partial GD
3. gonadal regression ovotesticular DSD |
Gonadal (ovarian) dysgenesis
1. ovotesticular DSD
2. testicular DSD
3. GD |
|
Disorder of androgen biosynthesis or action
1. Disorder of androgen biosynthesis
2. Defective androgen action
3. LH receptor defect
4. Defective action of AMH or AMH receptor |
Androgen excess
1. Fetal level (e.g. CAH)
2. Feto-placental level (e.g. Aromatase deficiency)
3. Maternal level (e.g. maternal ovarian or adrenal tumor) |
|
Others
(cloacal extrophy, severe hypospadias,…) |
Others
(cloacal extrophy, vaginal atresia, severe epispadias,…) |
DSD: disorder of sex development; GD: gonadal dysgenesis; CAH: congenital adrenal hyperplasia; AMH: anti Müllerian hormone.
This new terminology was initially strongly criticized, mainly by Arabic and African specialists: suffering from a "disorder" while not being really ill is not accepted in certain cultures, especially if the words "sex" or "sex development" are involved. Moreover, this classification has some additional limitations: The emphasis on the patient's karyotype may be irrelevant to patients, as well as from a clinical point of view. The proposed terminology does not refer to any of the functional or pathological characteristics of the dysgenetic gonad, nor does it group patients characterized by the same clinical phenotype. Therefore it is not considered a useful tool in daily clinical practice. At present, however, this new terminology and classification is maintained and seems to be increasingly accepted all over the world. Presently it is considered as a living document, avoiding the problems from the past and opening perspectives for amelioration. An overview of the revised nomenclature with regard to DSD is presented in Table 4.
Table 3. Overview of genes involved in 46, XY DSD and undervirilization or 46, XX DSD and overvirilization[4,5,7,18,22,23]
|
Condition |
Gene |
Adrenal
pathology |
Diagnostic features |
|
46, XY DSD with GD |
|
|
|
|
Smith-Lemli-Opitz |
DHCR7 |
+/- |
7 dehydrocholesterol↑ |
|
Leydig cell hypoplasia |
LHGCR |
- |
LH↑, response after hCG
stimulation↓ |
|
Congenital lipoid adrenal
hyperplasia |
STAR |
+ |
Disturbed biosynthesis of
all steroid hormones |
|
Cholesterol (P450) scc
deficiency |
CYP11A1 |
+ |
Disturbed biosynthesis of all steroid hormones |
|
3β HSD type II def |
HSD3B2 |
+ |
∆5:∆4 ratio↑, +/- MC insufficiency↑ |
|
17α-hydroxylase/17,20 lyase def |
CYP17 |
+ |
Pregnenolone↑, progesterone,11-deoxycorticosterone↑
17 OH-steroids↓, LH↑ |
|
P450 oxidoreductase def
|
POR
|
+
|
Features of 21-hydroxylase def, 17α-hydroxylase/17, 20 lyase def |
|
17β HSD type III
|
HSD17B3
|
-
|
T: androstenedion ratio <0.6 |
|
5α reductase type II def
|
SRD5A2
|
-
|
T: DHT ratio
(>20 after hCG stimulation)↑ |
|
AIS
|
AR
|
-
|
T and LH/FSH (variable)↑, AMH↑ |
|
46, XX DSD with androgen excess |
|
|
|
|
3β HSD type II def |
HSD3B2 |
+ |
ACTH↑, ∆5:∆4 ratio +/-
MC insufficiency↑ |
|
21-hydroxylase def |
CYP21A2 |
+ |
ACTH↑,17-OHP +/- MC
insufficiency↑ |
|
11 β-hydroxylase def |
CYP11B1 |
+ |
ACTH↑, 11-deoxycorticosterone↑,
11-deoxycortisol↑ |
|
P450 oxidoreductase def |
POR |
+ |
Features of 21-hydroxylase def, 17α-hydroxylase/ 17, 20 lyase def |
|
Aromatase def |
CYP19 |
- |
Androstendion↑, T↑, FSH/LH↑, oestrogens↓ |
|
Glucocorticoid resistance |
GRα |
- |
ACTH↑, 17-OHP↑, cortisol↑, MC↑, androgens↑, absent dexamethasone suppression |
Ssc: side chain cleavage; HSD: hydroxy steroid dehydrogenase; def: deficiency; MC: mineralocorticoïd; T: testosteron; DHT: dihydrotestosteron; OHP: hydroxyprogesteron; PCOS: polycystic ovary syndrome.
Table 4. Overview of the revised English nomenclature with regard to DSD[1,2]
|
Old terminology |
New terminology |
|
Intersex |
Disorder of sex development (DSD) |
|
Male pseudohermaphrodite |
46, XY DSD |
|
Undervirilisation of an XY male |
|
|
Undermasculinization of an XY male |
|
|
Female pseudohermaphrodite |
46, XX DSD |
|
Overvirilisation of an XX female |
|
|
Masculinization of an XX female |
|
|
True hermaphrodite |
Ovotesticular DSD |
|
XX male or XX sex reversal |
46, XX testicular DSD |
|
XY sex reversal |
46, XY complete gonadal
dysgenesis |
Gonadal phenotypes in DSD and their risk for the development of germ cell tumors
In 46, XX overvirilization syndromes, the gonad is always differentiated as an ovary, the source of androgens being the mother, the placenta or the fetal adrenals. 46, XY GD and sex chromosomal GD are characterized by extreme heterogeneity with regard to gonadal differentiation patterns. Our research results indicate that the earlier the gonadal developmental program is interrupted by a specific genetic defect, the less the gonad is able to differentiate into a testis or an ovary. The resulting gonadal histology is thus located somewhere on a gradual continuum with on the one end the well-differentiated testicle and on the other the normally differentiated ovary, depending on the underlying genetic defect (Fig. 2).[25,26] On this continuum, some distinct basic patterns can be clearly recognized: normal testis (eventually with maturation delay of germ cells), dysgenetic testis, primitive sex cords containing germ cells, streak fibrous tissue (without germ cells) with remnants of testicular or ovarian differentiation, as indicated by respectively SOX9 or FOXL2 positive areas, undifferentiated gonadal tissue, which has basically the same characteristics as streak tissue but which, importantly, contains isolated germ cells, and normal ovary (Fig. 3). Moreover, any combination of these patterns can be found within the same or the opposite gonad of the same patient. Thus other possibly micro environmental influences, e.g., the reach of a threshold, initiating a cascade of coupled reactions, may play a role, besides the genetic defect in a given patient. In 46, XY undervirilization syndromes, the gonad is differentiated as a testis, in which germ cells are usually delayed in their normal maturation process due to suboptimal support by the Sertoli cells. These germ cells usually survive for some years, but are prone to apoptosis, so that in most postpubertal gonads, the testis tubules are completely devoid of germ cells unless a tumor has emerged.[25,27-29]
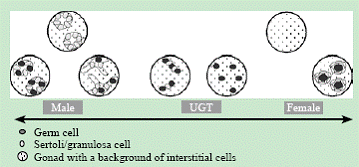
Fig. 2. The gonadal differentiation patterns that can be found in patients with gonadal dysgenesis do not appear strictly separated but may constitute a continuum, more or less differentiated into the male or female direction (modified from[26]).
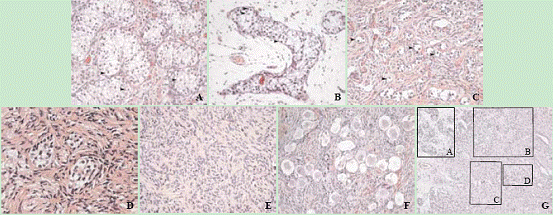
Fig. 3. A: Area with normal testis differentiation. The testis tubules contain germ cells (arrows). Patient karyotype: 45, X, inv(5) (q22q33.2); 46, X i(Y9), inv(5) (q22q33.2) (HE, original magnification × 200). B: Area with markedly dysgenetic testis tubules. The testis tubules contain germ cells (arrows). Same patient as A (HE, original magnification × 200). C: Area with undifferentiated gonadal tissue. Germ cells (arrows) line up with Sertoli/granulosa cells to form cord-like structures that are not differentiated into testis tubules, nor do they form ovarian follicular structures. Patient karyotype: 45, X/46, XY (HE, original magnification × 200). D: Testis tubules devoid of germ cells in a background of ovarian stroma. Patient karyotype 45, X/46, XY (HE, original magnification × 400). E: Area with streak gonadal tissue. Same patient as D. (HE, original magnification × 200). F: Ovarian differentiation pattern, showing germ cells enclosed in primordial follicles. Patient karyotype: 46, XX/47, XXY. (HE, original magnification × 200). G: Combination of various gonadal differentiation patterns within a limited area: A: testicular tissue; B: testis differentiation in a background of ovarian stroma; C: undifferentiated gonadal tissue; D: gonadoblastoma. Patient karyotype: 45, X/46, XY (HE, original magnification × 100).
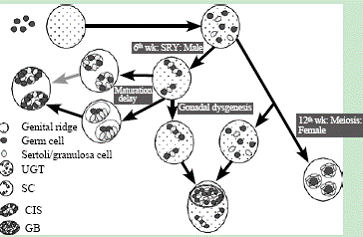
Fig. 4. Hypothesis for the development of CIS and GB in patients with DSD.[25] Model for the development of undifferentiated gonadal tissue, gonadoblastoma and carcinoma in situ in the dysgenetic gonad: Upper part: In the early embryo, germ cells migrate to the bipotential gonads and intermingle with pre-Sertoli/granulosa cells. Middle part: from the right to the left: After SRY expression during the 6th week of pregnany, pre-Sertoli cells and germ cells organize within primitive sex cords. These differentiate into seminiferous tubules under the influence of genes downstream of SRY. Specific pathological conditions cause a delay in normal germ cell development, leading to an increased risk for the development of carcinoma in situ. Lower part: In case SRY and/or the downstream cascade is not correctly activated due to a mutation in a gene involved in sex development, the gonad remains undifferentiated, or primitive sex cords do not develop further into seminiferous tubules. Since the activation of specific genes, necessary for proper ovarian differentiation also lacks, the gonad remains highly undifferentiated. Surviving germ cells within this gonad are at high risk for gonadoblastoma formation.
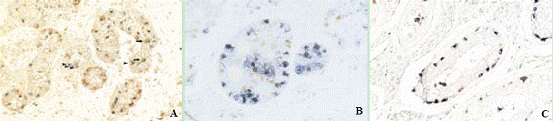
Fig. 5. OCT3/4 and TSPY expression in carcinoma in situ, gonadoblastoma and delayed maturation of germ cells. A: Delayed maturation versus malignant changes of germ cells in the testis: Germ cells (red staining, VASA) are normally located on the basal membrane in the postnatal testis, but in this testis, they are seen on the basal membrane as well as in the center of the tubules. OCT3/4 positive germ cells are normally only encountered in the fetal testis and are then located in the center of the tubules. In this testis, some germ cells have still not lost OCT3/4 expression (blue staining), they are located in the center of the tubules (arrows). Thus, these cells are delayed in their maturation, since they remain OCT3/4 positive until long after fetal life. However, some OCT3/4 positive germ cells are located on the basal membrane (arrow heads). Thus, these cells remain OCT3/4 positive after they made contact with the basal membrane and have to be considered as premalignant. It is to be expected that carcinoma in situ (and later an invasive tumor) would have developed later if gonadectomy had not been performed (OCT3/4-VASA double staining, original magnification × 400). B: OCT3/4 and TSPY co-expression in gonadoblastoma: Germ cells within the gonadoblastoma lesion express OCT3/4 (red) as well as TSPY (blue) (OCT3/4-TSPY double staining, original magnification × 200). C: OCT3/4 and TSPY co-expression in carcinoma in situ: Germ cells within carcinoma in situ express OCT3/4 (red) as well as TSPY (blue) (OCT3/4-TSPY double staining, original magnification × 200).
It is well known that germ cell tumors, specifically the non-invasive precursors carcinoma in situ (CIS) and gonadoblastoma (GB), and their invasive counterparts seminoma/dysgerminoma and non-seminoma, occur with increased frequency in patients with DSD and Y chromosomal material in their karyotype.[30-32] 46, XX overvirilized patients have no increased risk. Epidemiological data indicate that not the Y chromosome itself determines this risk, but the presence of a specific region on Y, called the "gonadoblastoma susceptibility region on the Y chromosome" (GBY region).[33-35] This hypothesis is supported by recent histological and molecular genetic research data, pointing at one strong candidate gene within this GBY region, the testis specific protein Y- encoded (TSPY).[36-39] TSPY is located on Yp, close to the centromeric region, some additional copies have also been identified on the proximal part of Yq, and thus this gene is situated, genetically spoken, very far from SRY, which is located on the distal part of Yp.
Immunohistochemical research has demonstrated that germ cells residing in a suboptimal environment, as seen in patients with GD or undervirilization syndromes, are characterized by a process of delayed maturation. Germ cells delayed in their maturation express OCT3/4 (alternatively called POU5F1), as malignant germ cells do. OCT3/4 is a transcription factor which is typically expressed in fetal, undifferentiated germ cells, but this expression is lost around or shortly after birth, thus, normally, postnatal germ cells do not express OCT3/4 any more. Germ cells in CIS and GB are strikingly positive for OCT3/4, this in combination with a very strong TSPY expression.[25, 28,40-42] The physiological role of OCT3/4 is most likely related to the survival of germ cells.[43] Moreover, oncogenic activity of OCT3/4 has been demonstrated in vitro in tumor cell lines derived from mice embryonic germ cells. The exact function of TSPY remains unknown, but it is believed to play a role in the mitotic proliferation of germ cells.[44] It is hypothesized that the observed combination of prolonged OCT3/4 expression and increased TSPY expression in the germ cells of DSD patients, is of pathogenetic relevance for the development of GCT in these patients.[27] At present, this hypothesis is further examined.[39]
The invasive seminoma (of the testis) and dysgerminoma (of the ovary) and non-seminoma represent the end stage invasive tumors arising from their non-invasive counterparts CIS and GB. Chromosomal characterization and expression analysis demonstrate that seminoma and dysgerminoma are indeed the same tumors.[45] CIS is only found in well differentiated testicular tissue, containing seminiferous tubules, whereas GB is found in undifferentiated gonadal tissue or in gonadal tissue characterized by only rudimentary testis differentiation (in which sex cords are found instead of seminiferous tubules).[25,27] The degree of gonadal differentiation is not only indicative of the type of precursor lesion (CIS or GB), but also determines the tumor risk: germ cell tumors arise mainly in highly undifferentiated gonadal tissue. This explains why an ovotestis (characterized by the presence of well-differentiated testicular tubules and ovarian follicles in the same individual) has a low risk for the development of GCT as compared to other forms of DSD (Fig. 4).[46]
Immunohistochemical techniques are used to study the degree of gonadal differentiation, as described above, but are also essential to perform a detailed study of the germ cells. Their number and degree of maturation are established, afterwards it is examined if their degree of maturation corresponds to their location within the seminiferous tubule (Fig. 5). This technique allows to differentiate between gonadal maturation delay on one hand and early CIS or GB on the other, which is otherwise not possible.[28] Consecutively, an estimation of the patient's risk for the development of an invasive GCT can be made according to the model as proposed in Fig. 6.[27] At present, this flowchart is based on a limited number of studies and relatively small patient series. However, if it turns out to be clinically useful, one could use this flowchart in the future to predict tumor risk in a specific patient on the basis of a gonadal biopsy. A limitation to this is the fact that one small biopsy in a young child is not necessarily representative for the whole gonad,[47] especially in the case of DSD.
The exact risk for the development of germ cell tumors in DSD patients is presently under debate. In older series, the incidence may vary between 5% and 75%. This is on one hand due to insufficient diagnostic tools to differentiate CIS from maturation delay, leading to important overdiagnosis of CIS, and on the other hand due to misclassification of clinical syndromes, based on the older, confusing nomenclature.[27,28] However, a recent review of the literature reveals some interesting data as described below:[27]
In cases of undervirilization, the mean incidence is estimated at 2.3%. However, according to the underlying diagnosis, there are important differences. In CAIS, the risk is extremely low, and since this is the most frequently encountered diagnosis, the mean incidence of undervirilization syndromes remains low. The risk is markedly higher in other diagnosis such as PAIS and 17β-HSD deficiency, but at present there are insufficient data to show a correct incidence of these rare syndromes (Table 5).
In the various undervirilization syndromes, the gonads are always well differentiated testes; therefore, only CIS occurs in this patient population, and GB is never encountered.
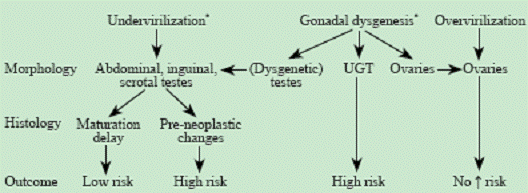 Fig. 6. Flowchart to estimate the risk for germ cell tumors in DSD patients.[27] The risk for the development of germ cell tumors is related to the degree and pattern of differentiation of the gonad. *: no risk in the absence of germ cells or in the absence of the TSPY gene in PBL and/or gonadal tissue. Fig. 6. Flowchart to estimate the risk for germ cell tumors in DSD patients.[27] The risk for the development of germ cell tumors is related to the degree and pattern of differentiation of the gonad. *: no risk in the absence of germ cells or in the absence of the TSPY gene in PBL and/or gonadal tissue.
It is hypothesized that exposure to androgens during and after puberty stimulates the development of GCT in male DSD patients. However, the underlying mechanism remains to be elucidated (Germ cells do not express the androgen receptor; possibly, a Sertoli cell mediated mechanism is involved).
In patients with GD, the mean risk is estimated at 12%. GB is mostly derived from undifferentiated gonadal tissue or primitive sex cords. However, CIS can also be encountered if some testicular differentiation is found within the gonad.[25]
In undervirilization the risk in GD is probably dependent on the underlying diagnosis, but the scarce data do not allow an estimation of the risk per diagnostic category or per gene defect (Table 5).
GB and invasive tumors in patients with GD occur at a considerably younger age than CIS does in patients with undervirilization syndromes. This finding indicates that pubertal hormones probably play a minor or no role at all in this context, since most GB is found before the beginning of puberty. Moreover, very few or no hormonal production takes place in undifferentiated gonadal tissue.
Conclusion
In recent years, considerable progress has been made in our knowledge and management of DSD.
In the first place, a number of hitherto unknown genes involved in gonadal differentiation have been described (e.g., SF1, DAX1, RSPO1). Molecular genetic research becomes increasingly available, allowing a straightforward diagnosis more frequently.
Secondly, extensive research has been done on the development of GCT in DSD patients. This opens perspectives to estimate tumor risk in the individual patient according to the result of a gonadal biopsy, provided that the biopsy is analyzed in a specialized reference center, which led, in our center at least, to a more conservative approach with regard to gonadectomy in a number of cases. An important limitation to the application of this technique on a wide scale is the fact that a gonadal biopsy in a small child with DSD is probably insufficiently representative for the whole gonad.
Finally, the 2005 international consensus meeting has led to a new nomenclature and certainly also to an increased attention for the psychological problems of DSD patients. A renewed interest to cooperate in large scale international research protocols is noticed, and the problems of DSD patients have now been recognized by a number of European governmental organizations, leading to new funding possibilities for research on this topic.
As a consequence, it is expected that in the near future, this will lead to considerable changes in the management of patients with DSD, for example, an increased emphasis on a multidisciplinary approach of DSD patients, a more frequent choice for the male gender, even in cases of severe undervirilization, a more conservative approach with regard to gonadectomy (e.g., in CAIS and ovotesticular DSD), and an attempt to maximize fertility chances in some patients by the application of new cryopreservation techniques at a very young age.
Funding: Cools M is granted by the Flanders Research Foundation (FWO).
Ethical approval: Not needed.
Competing interest: All authors declare that they have no competing interests to disclose.
Contributors: All the authors declare that they contributed to the concept and design of this article, to the acquisition, analysis and interpretation of data, to the article draft and its critical revision and to the final approval of the submitted version.
References
1 Hughes IA, Houk C, Ahmed SF, Lee PA; LWPES Consensus Group; ESPE Consensus Group. Consensus statement on management of intersex disorders. Arch Dis Child 2006;91:554-563.
2 Lee PA, Houk CP, Ahmed SF, Hughes IA; International Consensus Conference on Intersex organized by the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. Consensus statement on management of intersex disorders. International Consensus Conference on Intersex. Pediatrics 2006;118:e488-500.
3 Brennan J, Capel B. One tissue, two fates: molecular genetic events that underlie testis versus ovary development. Nat Rev Genet 2004;5:509-521.
4 Grumbach MM, Hughes IA, Conte FA. Disorders of sex differentiation. In: Larsen PR, Kronenberg HM, Melmed S, Polonsky KM, eds. Williams textbook of endocrinology, 10th ed. Philadelphia: W.B. Saunders (Elsevier), 2003: 842-1002.
5 Vaiman D, Pailhoux E. Mammalian sex reversal and intersexuality: deciphering the sex-determination cascade. Trends Genet 2000;16:488-494.
6 Kildal W, Kraggerud SM, Abeler VM, Heim S, Trope CG, Kristensen GB, et al. Genome profiles of bilateral dysgerminomas, a unilateral gonadoblastoma, and a metastasis from a 46, XY phenotypic female. Hum Pathol 2003;34:946-949.
7 Fleming A, Vilain E. The endless quest for sex determination genes. Clin Genet 2005;67:15-25.
8 Achermann JC, Meeks JJ, Jameson JL. Phenotypic spectrum of mutations in DAX-1 and SF-1. Mol Cell Endocrinol 2001;185:17-25.
9 Wylie C. Germ cells. Curr Opin Genet Dev 2000;10:410-413.
10 Wilhelm D, Palmer S, Koopman P. Sex determination and gonadal development in mammals. Physiol Rev 2007;87:1-28.
11 Parma P, Radi O, Vidal V, Chaboissier MC, Dellambra E, Valentini S, et al. R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat Genet 2006;38:1304-1309.
12 Kim Y, Kobayashi A, Sekido R, DiNapoli L, Brennan J, Chaboissier MC, et al. Fgf9 and Wnt4 act as antagonistic signals to regulate mammalian sex determination. PLoS Biol 2006;4:e187.
13 Kobayashi A, Behringer RR. Developmental genetics of the female reproductive tract in mammals. Nat Rev Genet 2003;4:969-980.
14 Anderson RA, Sharpe RM. Regulation of inhibin production in the human male and its clinical applications. Int J Androl 2000;23:136-144.
15 Ferlin A, Foresta C. Insulin-like factor 3: a novel circulating hormone of testicular origin in humans. Ann N Y Acad Sci 2005;1041:497-505.
16 Biason-Lauber A, Konrad D, Navratil F, Schoenle EJ. A WNT4 mutation associated with Mullerian-duct regression and virilization in a 46,XX woman. N Engl J Med 2004;351:792-798.
17 Miller WL. Disorders of androgen biosynthesis. Semin Reprod Med 2002;20:205-216.
18 Kim CJ, Lin L, Huang N, Quigley CA, AvRuskin TW, Achermann JC, et al. Severe combined adrenal and gonadal deficiency caused by novel mutations in the cholesterol side chain cleavage enzyme, P450scc. J Clin Endocrinol Metab 2008;93:696-702.
19 Quigley CA, De Bellis A, Marschke KB, el-Awady MK, Wilson EM, French FS. Androgen receptor defects: historical, clinical, and molecular perspectives. Endocr Rev 1995;16:271-321.
20 Hannema SE, Scott IS, Hodapp J, Martin H, Coleman N, Schwabe JW, et al. Residual activity of mutant androgen receptors explains wolffian duct development in the complete androgen insensitivity syndrome. J Clin Endocrinol Metab 2004;89:5815-5822.
21 Boehmer AL, Brinkmann O, Bruggenwirth H, van Assendelft C, Otten BJ, Verleun-Mooijman MC, et al. Genotype versus phenotype in families with androgen insensitivity syndrome. J Clin Endocrinol Metab 2001;86:4151-4560.
22 Ahmed SF, Hughes IA. The genetics of male undermasculinization. Clin Endocrinol (Oxf) 2002;56:1-18.
23 Hiort O, Holterhus PM. The molecular basis of male sexual differentiation. Eur J Endocrinol 2000;142:101-110.
24 Houk CP, Lee PA. Intersexed states: diagnosis and management. Endocrinol Metab Clin North Am 2005;34: 791-810, xi.
25 Cools M, Stoop H, Kersemaekers AM, Drop SL, Wolffenbuttel KP, Bourguignon JP, et al. Gonadoblastoma arising in undifferentiated gonadal tissue within dysgenetic gonads. J Clin Endocrinol Metab 2006;91:2404-2413.
26 Hersmus R, de Leeuw BH, Wolffenbuttel KP, Drop SL, Oosterhuis JW, Cools M, et al. New insights into type II germ cell tumor pathogenesis based on studies of patients with various forms of disorders of sex development (DSD). Mol Cell Endocrinol 2008;291:1-10.
27 Cools M, Drop SL, Wolffenbuttel KP, Oosterhuis JW, Looijenga LH. Germ cell tumors in the intersex gonad: old paths, new directions, moving frontiers. Endocr Rev 2006;27:468-484.
28 Cools M, van Aerde K, Kersemaekers AM, Boter M, Drop SL, Wolffenbuttel KP, et al. Morphological and immunohistochemical differences between gonadal maturation delay and early germ cell neoplasia in patients with undervirilization syndromes. J Clin Endocrinol Metab 2005;90:5295-5303.
29 Looijenga LH, Hersmus R, Oosterhuis JW, Cools M, Drop SL, Wolffenbuttel KP. Tumor risk in disorders of sex development (DSD). Best Pract Res Clin Endocrinol Metab 2007;21:480-495.
30 Manuel M, Katayama PK, Jones HW Jr. The age of occurrence of gonadal tumors in intersex patients with a Y chromosome. Am J Obstet Gynecol 1976;124:293-300.
31 Scully RE. Gonadoblastoma. A review of 74 cases. Cancer 1970;25:1340-1356.
32 Verp MS, Simpson JL. Abnormal sexual differentiation and neoplasia. Cancer Genet Cytogenet 1987;25:191-218.
33 Tsuchiya K, Reijo R, Page DC, Disteche CM. Gonadoblastoma: molecular definition of the susceptibility region on the Y chromosome. Am J Hum Genet 1995;57:1400-1407.
34 Salo P, Kaariainen H, Petrovic V, Peltomaki P, Page DC, de la Chapelle A. Molecular mapping of the putative gonadoblastoma locus on the Y chromosome. Genes Chromosomes Cancer 1995;14:210-214.
35 Page DC. Hypothesis: a Y-chromosomal gene causes gonadoblastoma in dysgenetic gonads. Development 1987;101 Suppl:151-155.
36 Lau YF. Gonadoblastoma, testicular and prostate cancers, and the TSPY gene. Am J Hum Genet 1999;64:921-927.
37 Lau Y, Chou P, Iezzoni J, Alonzo J, Komuves L. Expression of a candidate gene for the gonadoblastoma locus in gonadoblastoma and testicular seminoma. Cytogenet Cell Genet 2000;91:160-164.
38 Kersemaekers AM, Honecker F, Stoop H, Cools M, Molier M, Wolffenbuttel K, et al. Identification of germ cells at risk for neoplastic transformation in gonadoblastoma. Hum Pathol 2005;36:512-521.
39 Schnieders F, Dork T, Arnemann J, Vogel T, Werner M, Schmidtke J. Testis-specific protein, Y-encoded (TSPY) expression in testicular tissues. Hum Mol Genet 1996;5:1801-1807.
40 Cools M, Honecker F, Stoop H, Veltman JD, de Krijger RR, Steyerberg E, et al. Maturation delay of germ cells in trisomy 21 fetuses results in increased risk for the development of testicular germ cell tumors. Hum Pathol 2006;37:101-111.
41 Honecker F, Stoop H, de Krijger RR, Chris Lau YF, Bokemeyer C, Looijenga LH. Pathobiological implications of the expression of markers of testicular carcinoma in situ by fetal germ cells. J Pathol 2004;203:849-857.
42 Stoop H, Honecker F, Cools M, de Krijger R, Bokemeyer C, Looijenga LH. Differentiation and development of human female germ cells during prenatal gonadogenesis: an immunohistochemical study. Hum Reprod 2005;20:1466-1476.
43 Kehler J, Tolkunova E, Koschorz B, Pesce M, Gentile L, Boiani M, et al. Oct4 is required for primordial germ cell survival. EMBO Rep 2004;5:1078-1083.
44 Schnieders F, Dork T, Arnemann J, Vogel T, Werner M, Schmidtke J. Testis-specific protein, Y-encoded (TSPY) expression in testicular tissues. Hum Mol Genet 1996;5:1801-1807.
45 Looijenga LH, Hersmus R, Gillis AJ, Pfundt R, Stoop HJ, van Gurp RJ, et al. Genomic and expression profiling of human spermatocytic seminomas: primary spermatocyte as tumorigenic precursor and DMRT1 as candidate chromosome 9 gene. Cancer Res 2006;66:290-302.
46 van Niekerk WA, Retief AE. The gonads of human true hermaphrodites. Hum Genet 1981;58:117-122.
47 Berthelsen JG, Skakkebaek NE. Value of testicular biopsy in diagnosing carcinoma in situ testis. Scand J Urol Nephrol 1981;15:165-168.
Received July 24, 2008 Accepted after revision February 6, 2009
|
